Transforms RNA-sequencing data into actionable clinical insights with automated reports.
![]()
![]()
Documentation | umccr.github.io/RNAsum
What is RNAsum?
RNAsum is an R package that integrates whole-genome sequencing (WGS) and whole-transcriptome sequencing (WTS) data to generate comprehensive, interactive HTML reports for cancer patient samples.
Quick start
RNAsum can be installed using one of the following two methods.
Installation
Option 1: from GitHub
RNAsum depends on pdftools, which requires system-level libraries (poppler, cairo, etc.) to be installed before installing the R package.
System dependencies installation
Ubuntu/Debian:
sudo apt-get install libpoppler-cpp-dev libharfbuzz-dev libfribidi-dev \
libfreetype6-dev libcairo2-dev libpango1.0-devmacOS:
HPC/Cluster (without root):
If you do not have root access (e.g., on a cluster), creating a fresh Conda environment is the most reliable way to provide necessary system libraries:
Once system dependencies are met, you can install the package directly from GitHub from within R console.
# 1. Increase timeout to prevent download failure for RNAsum.data
options(timeout = 600)
# 2. Install via remotes
if (!require("remotes")) install.packages("remotes")
remotes::install_github("umccr/RNAsum")Workflow
The pipeline consists of five main components.
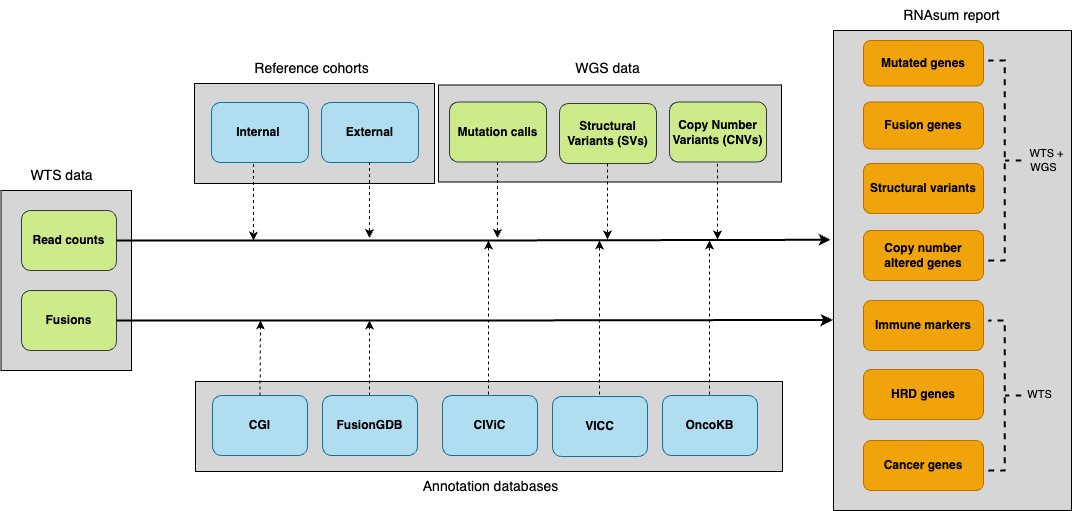
- WTS data collection: ingests per-gene read counts and gene fusions.
- Reference integration: normalises against reference cohorts.
- WGS data integration: links genomic alterations with expression data.
- Knowledge enrichment: annotates with clinically relevant databases.
- Report generation: prioritises findings and creates interactive visualizations.
Usage
Add RNAsum to PATH environment variable.
rnasum_cli=$(Rscript -e 'cat(system.file("cli", package="RNAsum"))')
ln -sf "$rnasum_cli/rnasum.R" "$rnasum_cli/rnasum"
export PATH="$rnasum_cli:$PATH"Batch effect considerations
When comparing clinical RNA-seq samples to TCGA reference cohorts, batch effects can significantly impact expression rankings due to protocol differences (e.g., ribo-depletion vs poly-A selection, different library preparation methods).
RNAsum provides the --batch_rm parameter to address these differences:
When to use --batch_rm:
- Clinical samples use different RNA-seq protocols than TCGA
- An internal reference cohort was processed with the same protocol
- Expression rankings appear systematically skewed
Alternative approaches:
- Use tissue-matched internal reference cohorts when available
- Consider protocol-specific thresholds for expression classification
- Validate findings with orthogonal methods when possible
Common options
| Option | Description | Default |
|---|---|---|
--sample_name |
Sample identifier | Required |
--dataset |
TCGA reference cohort | PANCAN |
--batch_rm |
Remove batch effects | FALSE |
--salmon |
Salmon quantification file | - |
--kallisto |
Kallisto abundance file | - |
--arriba_tsv |
Arriba fusion detection output | - |
--pcgr_tiers_tsv |
PCGR variant calls (tier 1-4) | - |
--cn_gene_tsv |
Copy number by gene | - |
--filter |
Filter low-expressed genes | TRUE |
Run rnasum --help to get complete list of options.
For format and minimal content of input files (e.g. --pcgr_tiers_tsv, --cn_gene_tsv, --sv_tsv), see Input file formats.
Note: human reference genome GRCh38 (Ensembl based annotation version 105) is used for gene annotation by default. GRCh37 is no longer supported.
Examples
Test data: in /inst/rawdata/test_data folder of the GitHub repo
Runtime: < 15 minutes (16GB RAM, 1 CPU)
Scenario 1: WGS + WTS (recommended)
Comprehensive reporting, in which WGS-based findings are used as a primary source for expression profile prioritisation.
cd $rnasum_cli
rnasum \
--sample_name test_sample_WTS \
--dataset TEST \
--salmon "$PWD/../rawdata/test_data/dragen/TEST.quant.genes.sf" \
--arriba_pdf "$PWD/../rawdata/test_data/dragen/arriba/fusions.pdf" \
--arriba_tsv "$PWD/../rawdata/test_data/dragen/arriba/fusions.tsv" \
--dragen_fusions "$PWD/../rawdata/test_data/dragen/test_sample_WTS.fusion_candidates.final" \
--pcgr_tiers_tsv "$PWD/../rawdata/test_data/small_variants/TEST-snvs_indels.tiers.tsv" \
--cn_gene_tsv "$PWD/../rawdata/test_data/copy_number/TEST.cnv.gene.tsv" \
--sv_tsv "$PWD/../rawdata/test_data/structural/TEST-sv.tsv" \
--report_dir "$PWD/../rawdata/test_data/RNAsum" \
--save_tables FALSE \
--filter TRUEThe HTML report test_sample_WTS.RNAsum.html will be created in the inst/rawdata/test_data/dragen/RNAsum folder.
Scenario 2: WTS only
Basic reporting including information about detected gene fusions and expression levels of key genes.
cd $rnasum_cli
rnasum \
--sample_name test_sample_WTS \
--dataset TEST \
--salmon "$PWD/../rawdata/test_data/dragen/TEST.quant.genes.sf" \
--arriba_pdf "$PWD/../rawdata/test_data/dragen/arriba/fusions.pdf" \
--arriba_tsv "$PWD/../rawdata/test_data/dragen/arriba/fusions.tsv" \
--report_dir "$PWD/../rawdata/test_data/RNAsum" \
--save_tables FALSE \
--filter TRUEThe HTML report test_sample_WTS.RNAsum.html will be created in the inst/rawdata/test_data/dragen/RNAsum folder.
Batch effects assessment
Assess potential batch effects between the clinical sample and TCGA reference data before running the main analysis. This is particularly important when using different RNA-seq protocols.
Basic setup
The batch assessment functions (assess_batch_effects(), quick_batch_check()) are exported by RNAsum starting from the current release, so once the package is installed/upgraded user can call them directly after library(RNAsum) — no source() is needed.
If user cannot upgrade and are working from a local clone of the source tree, the user can instead source the file directly from the repository:
# Only needed when working from a local source clone and not from an installed RNAsum
# source("R/batch_assessment.R")Quick start
library(RNAsum)
# Load test sample data (Salmon gene quantification)
test_file <- system.file("rawdata/test_data/dragen/TEST.quant.genes.sf", package = "RNAsum")
sample_data <- read.delim(test_file)
sample_tpm <- setNames(sample_data$TPM, sample_data$Name)
# Get reference data
ref_paths <- get_refdata(dataset = "TEST", batch_rm = FALSE)
ref_counts <- utils::read.table(gzfile(ref_paths$ext_ref$counts),
header = TRUE, sep = "\t", row.names = NULL)
ref_matrix <- as.matrix(ref_counts[, -1])
rownames(ref_matrix) <- ref_counts[[1]]
# Quick assessment using most variable genes (default)
batch_results <- assess_batch_effects(
sample_data = sample_tpm,
reference_data = ref_matrix,
gene_set_type = "top_n",
n_genes = 2000,
output_dir = "batch_assessment_results"
)Assessment interpretation:
-
PCA distance percentile > 95%: High batch effect risk - consider
--batch_rm - Median correlation < 0.7: Substantial protocol differences detected
- Extreme Z-scores > 10%: Expression ranking issues likely
Detailed gene set options and examples
RNAsum provides three gene set options for targeted batch assessment.
Option 1: Top variable genes (default)
# Standard assessment using most variable genes (recommended for general use)
batch_results <- assess_batch_effects(
sample_data = sample_tpm,
reference_data = ref_matrix,
gene_set_type = "top_n", # Uses most variable genes
n_genes = 2000, # Number of genes to analyze
protocol_clinical = "ribo-depletion",
protocol_reference = "TCGA_poly-A",
output_dir = "batch_assessment_topn"
)Option 2: Cancer gene set
# Combined cancer genes database (1315 unique genes)
batch_cancer_combined <- assess_batch_effects(
sample_data = sample_tpm,
reference_data = ref_matrix,
gene_set_type = "cancer_genes",
cancer_gene_source = "combined", # Combined UMCCR + OncoKB databases
protocol_clinical = "ribo-depletion",
protocol_reference = "TCGA_poly-A",
output_dir = "batch_assessment_cancer_combined"
)
# Quick assessment with cancer genes
quick_batch_check(
sample_data = sample_tpm,
reference_data = ref_matrix,
gene_set_type = "cancer_genes",
cancer_gene_source = "combined",
save_plots = TRUE
)Option 3: Custom gene set
# User-defined gene sets (genes must match sample data format)
# Note: Ensure gene IDs match data format (Ensembl IDs vs gene symbols)
# Method 1: Direct Ensembl ID specification
custom_genes_direct <- c("ENSG00000141510", "ENSG00000012048", "ENSG00000139618") # TP53, BRCA1, BRCA2
batch_custom_direct <- assess_batch_effects(
sample_data = sample_tpm,
reference_data = ref_matrix,
gene_set_type = "custom",
gene_subset = custom_genes_direct,
protocol_clinical = "ribo-depletion",
protocol_reference = "TCGA_poly-A",
output_dir = "batch_assessment_custom_direct"
)
# Method 2: Use cancer genes with automatic conversion (recommended)
batch_cancer_symbols <- assess_batch_effects(
sample_data = sample_tpm,
reference_data = ref_matrix,
gene_set_type = "cancer_genes", # Enhanced: handles symbols automatically
cancer_gene_source = "combined", # 1315 genes with auto-conversion
protocol_clinical = "ribo-depletion",
protocol_reference = "TCGA_poly-A",
output_dir = "batch_assessment_cancer_auto"
)Prerequisites for cancer gene sets (Option 2)
For cancer gene set analysis, use the enhanced built-in functionality with automatic format conversion:
- No external files needed - Cancer gene databases and format conversion are built-in
- Automatic gene ID conversion - Handles Ensembl IDs ↔︎ gene symbols transparently
- Three cancer databases available - UMCCR (1248), OncoKB (1019), Combined (1315 genes)
- Smart format detection - Automatically detects and converts incompatible formats
Complete workflow example
Running all three options with TEST data
# Streamlined batch assessment workflow using enhanced functionality
library(RNAsum)
# Load test sample data
test_file <- system.file("rawdata/test_data/dragen/TEST.quant.genes.sf", package = "RNAsum")
sample_data <- read.delim(test_file)
sample_tpm <- setNames(sample_data$TPM, sample_data$Name)
# Get reference data
ref_paths <- get_refdata(dataset = "TEST", batch_rm = FALSE)
ref_counts <- utils::read.table(gzfile(ref_paths$ext_ref$counts),
header = TRUE, sep = "\t", row.names = NULL)
ref_matrix <- as.matrix(ref_counts[, -1])
rownames(ref_matrix) <- ref_counts[[1]]
# OPTION 1: Top variable genes
batch_topn <- assess_batch_effects(
sample_data = sample_tpm,
reference_data = ref_matrix,
gene_set_type = "top_n",
n_genes = 2000,
protocol_clinical = "ribo-depletion",
protocol_reference = "TCGA_poly-A"
)
# OPTION 2: Cancer genes
batch_cancer <- assess_batch_effects(
sample_data = sample_tpm,
reference_data = ref_matrix,
gene_set_type = "cancer_genes",
cancer_gene_source = "combined",
protocol_clinical = "ribo-depletion",
protocol_reference = "TCGA_poly-A"
)
# OPTION 3: Custom genes
custom_ensembl <- c("ENSG00000141510", "ENSG00000012048", "ENSG00000139618") # TP53, BRCA1, BRCA2
batch_custom <- assess_batch_effects(
sample_data = sample_tpm,
reference_data = ref_matrix,
gene_set_type = "custom",
gene_subset = custom_ensembl,
protocol_clinical = "ribo-depletion",
protocol_reference = "TCGA_poly-A"
)
# Compare results
cat("=== Enhanced Batch Assessment Results ===\n")
cat("Top genes analysis: ", round(batch_topn$pca_results$distance_percentile * 100, 1), "% PCA distance percentile\n")
cat("Cancer genes analysis: ", round(batch_cancer$pca_results$distance_percentile * 100, 1), "% PCA distance percentile\n")
cat("Custom genes analysis: ", round(batch_custom$pca_results$distance_percentile * 100, 1), "% PCA distance percentile\n")What’s in the report?
RNAsum generates an interactive HTML report with the following core sections:
- Findings summary: summary of genes listed across various report sections
- Mutated genes: expression of genes with somatic mutations (requires WGS)
- Fusion genes: detected gene fusions with functional annotations
- Structural variants: expression of genes located within structural variants (requires WGS)
- CN altered genes: expression in CN-gained/lost regions (requires WGS)
- Cancer genes: expression of cancer-associated genes
Available reference datasets
RNAsum includes 33 TCGA cancer type cohorts for comparative analysis:
| Cancer Type | Dataset Code | Samples |
|---|---|---|
| Pan-Cancer | PANCAN |
330 |
| Breast Invasive Carcinoma | BRCA |
300 |
| Lung Adenocarcinoma | LUAD |
300 |
| Pancreatic Adenocarcinoma | PAAD |
150 |
See the complete TCGA projects summary table.
Documentation
| Resource | Link |
|---|---|
| Full documentation | umccr.github.io/RNAsum |
| Workflow details | Workflow details |
| Batch assessment guide | Batch assessment |
| Report structure | Report structure |
| TCGA datasets | TCGA projects summary |
Contributing
We welcome contributions! Please see our Code of Conduct and contribution guidelines.
Reporting Issues
Found a bug or have a feature request? Open an issue.
Citation
If you use RNAsum please cite:
Kanwal S, Marzec J, Diakumis P, Hofmann O, Grimmond S (2024). “RNAsum: An R package to comprehensively post-process, summarise and visualise genomics and transcriptomics data.” version 1.1.0, https://umccr.github.io/RNAsum/
A BibTeX entry for LaTeX users is
@Unpublished{,
title = {RNAsum: An R package to comprehensively post-process, summarise and visualise genomics and transcriptomics data},
author = {Sehrish Kanwal and Jacek Marzec and Peter Diakumis and Oliver Hofmann and Sean Grimmond},
year = {2024},
note = {version 1.1.0},
url = {https://umccr.github.io/RNAsum/},
}